Benchmarking Hydrogen and Carbon NMR Chemical Shifts at HF, DFT, and MP2 Levels
23-Jan-2014
J. Chem. Theory Comput., 2014, DOI: 10.1021/ct400780f, 10 (2), pp 572–578 published on 23.01.2014
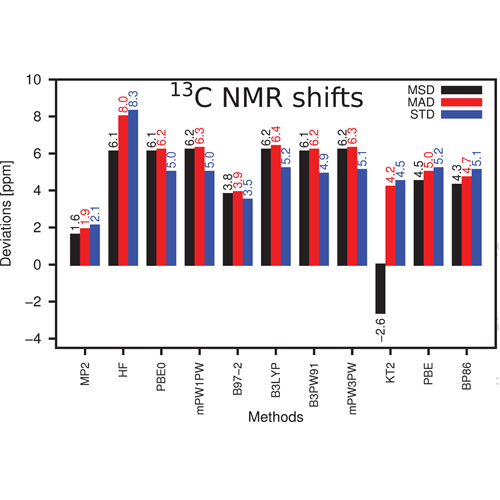
An extensive study of error distributions for calculating hydrogen and carbon NMR chemical shifts at Hartree–Fock (HF), density functional theory (DFT), and Møller–Plesset second-order perturbation theory (MP2) levels is presented. Our investigation employs accurate CCSD(T)/cc-pVQZ calculations for providing reference data for 48 hydrogen and 40 carbon nuclei within an extended set of chemical compounds covering a broad range of the NMR scale with high relevance to chemical applications, especially in organic chemistry. Besides the approximations of HF, a variety of DFT functionals, and conventional MP2, we also present results with respect to a spin component-scaled MP2 (GIAO-SCS-MP2) approach. For each method, the accuracy is analyzed in detail for various basis sets, allowing identification of efficient combinations of method and basis set approximations.